来源: 时间:2022-01-15 15:35:09
人类中有许多 “罕见” 的遗传疾病,被归类为这种疾病,因为很少有人受到这种疾病的影响。溶酶体贮积障碍在其中尤为突出。他们中的许多人无法治愈,因为制药公司对投资开发针对这些疾病的药物几乎没有兴趣。政府对这些疾病研究的支持也很有限。
虽然患有任何一种溶酶体贮积障碍的人数相当低,但它们加在一起占了大量患者。仅在印度,据估计有数百万人患有这些 “罕见” 疾病。
在研究人体细胞中的硫代谢时,我们被引入了一种称为cystinosis的疾病,该疾病现已成为我们研究的主要重点之一。
在正常的人类细胞中,蛋白质在溶酶体内分解成氨基酸,溶酶体是细胞内的几个亚结构之一,然后借助特定的转运蛋白移出。Cystinosin是一种转运蛋白,可将胱氨酸 (含硫氨基酸 “半胱氨酸” 的氧化形式) 从溶酶体中泵入细胞质。
由于遗传突变,cystinosin转运蛋白在cystinosis中变得无功能,因此胱氨酸无法移出并被困在溶酶体中。
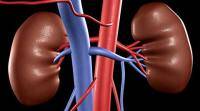
高浓度的胱氨酸会导致其在每个体内细胞的溶酶体内结晶,从而影响其正常功能。这种疾病表现为发育迟缓,视力丧失和肾脏功能障碍。
这种疾病的问题是,它不容易被诊断出来。理想情况下,当父母第一次抱怨孩子缺乏成长时,应该由儿科医生诊断。但大多数情况下,尤其是在我们国家,直到肾脏停止功能后才被发现。在那个阶段,医生可以提供的唯一补救措施是肾脏移植。
在早期发现的情况下,可以给患者服用一种称为半胱胺的药物,但必须每六个小时服用一次。这是一种痛苦的治疗方法,在印度每年花费约40万卢比,即使这样,患者的寿命也不会超过30-35岁,尽管比没有药物治疗的患者通常要多得多。
在印度,马德拉斯骨科和创伤研究所 (MIOT) 肾脏病学负责人Rajan Ravichandran博士对这种疾病做了一些开创性的工作,不仅在治疗上,而且在编目和研究上。
他在印度建立了一个半胱氨酸病患者登记处。这个注册表现在大约有25个名字。通过他创立的印度cystinosis分会和一个健康基金会,他还支持印度许多贫困患者的治疗费用。
与ravichandra博士密切合作,我们一直在对这种疾病进行研究,并开发与印度人口相关的突变图。
我们一直在探索多个角度来了解这种疾病及其原因。其中之一是了解胱氨酸无法退出溶酶体的初始生化后果。这种生化知识可能有助于解释为什么主要是肾脏受到影响。
我们的研究使我们能够提出一个假设来解释溶酶体中胱氨酸积累后的一些事件。我们相信,这可以让我们对这种疾病有一个好的、新的洞察力。我们还在研究高胱氨酸水平如何与细胞 “对话”。为此,我们一直在从外部注入大量这种氨基酸,以查看细胞的行为。
在由美国Cystinosis研究基金会资助的实验中,我们一直在使用面包酵母作为我们的模型生物,面包酵母是用于酿酒和烘焙的酵母。这种单细胞酵母与多细胞人类有很大的不同,酵母菌没有肾脏,也不会患有半胱氨酸病。然而,关键是两种生物都是 “真核生物”,因此具有相似的细胞内结构和生化途径。因此,使用该模型生物来研究cystinosin功能的关键方面是证明人类cystinosin蛋白 “放入” 该生物中时,其功能与其原始形式相似。
这种成功开发的酵母模型将进一步帮助我们了解突变对这种蛋白质功能的影响以及这种疾病的生化基础。
这项研究是由Anand Bachawat和team IISER,Mohali进行的
相关推荐
猜你喜欢